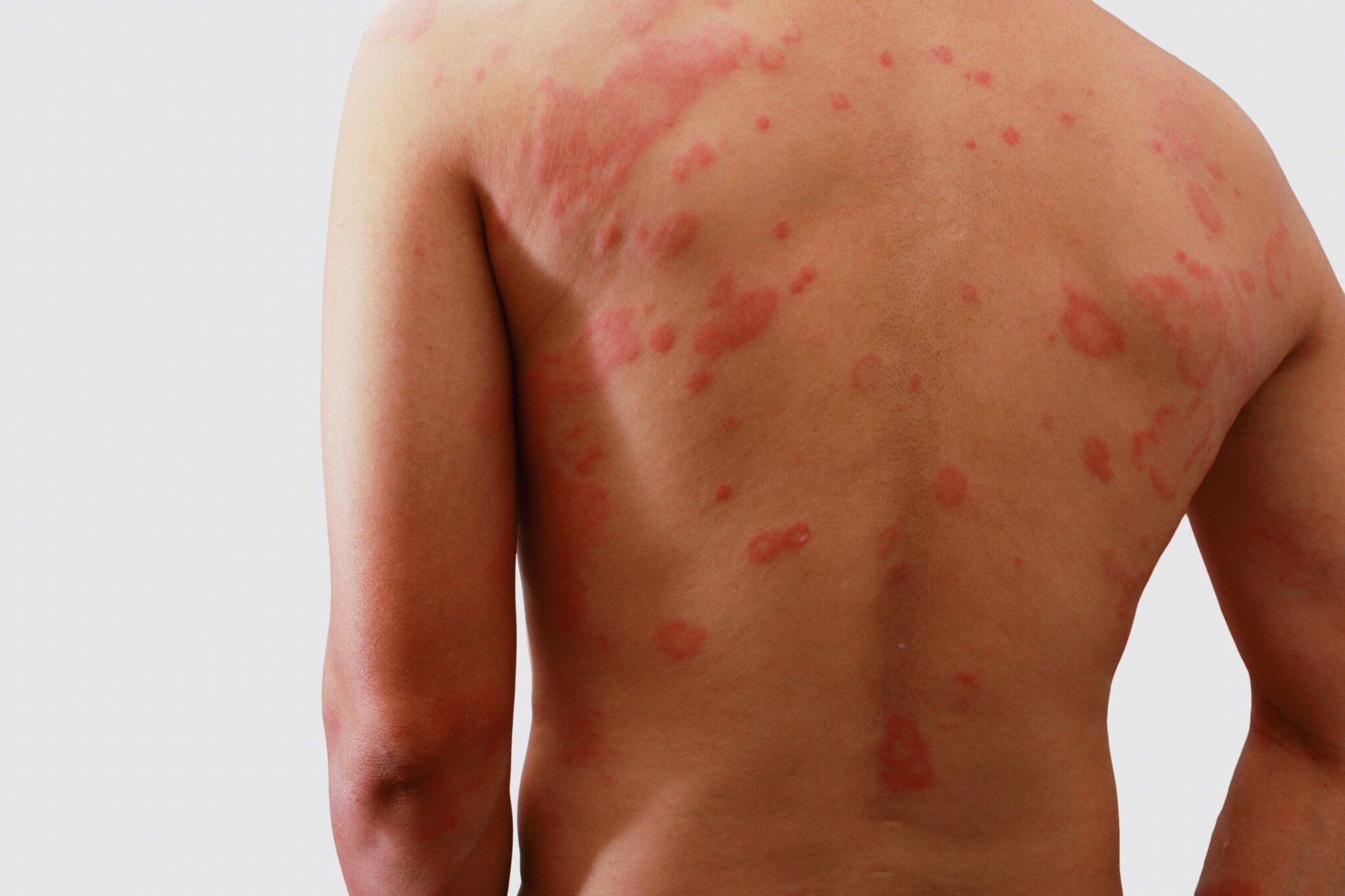
Zespół Stevensa-Johnsona (SJS) to rzadka, ale niezwykle poważna, zagrażająca życiu reakcja nadwrażliwości, która atakuje skórę i błony śluzowe. Charakteryzuje się rozległymi zmianami skórnymi przypominającymi oparzenia, a także zajęciem błon śluzowych jamy ustnej, oczu, narządów płciowych i dróg oddechowych. Stanowi on mniej nasiloną formę spektrum chorobowego, którego najcięższym biegunem jest zespół toksycznej nekrolizy (TEN), znany również jako Zespół Lyella. Chociaż obie jednostki chorobowe różnią się stopniem zajęcia powierzchni ciała, ich etiologia, patogeneza i leczenie są bardzo podobne.
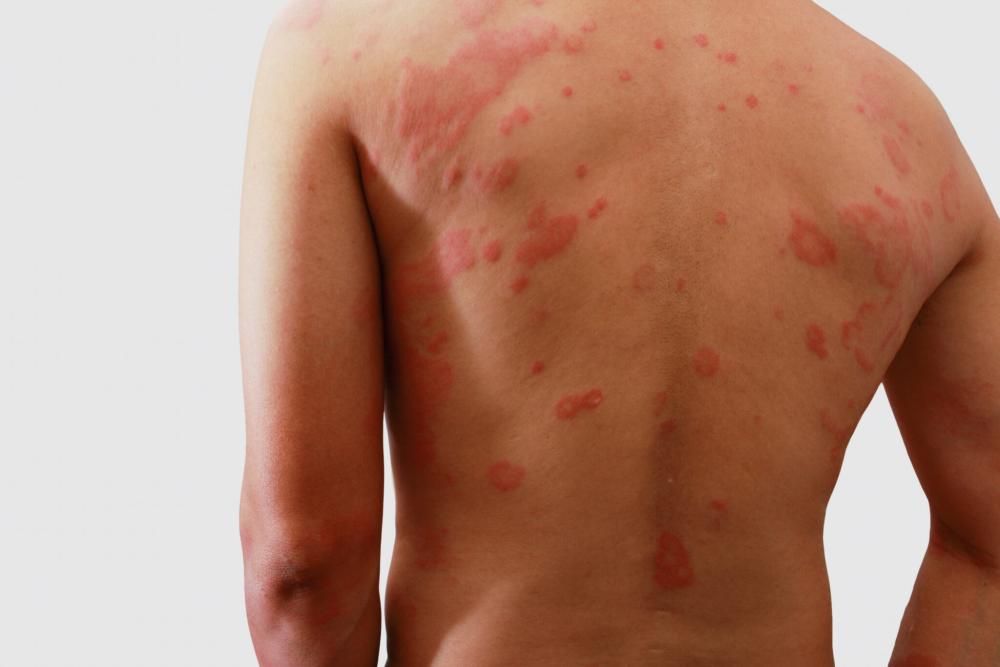
Definicja i klasyfikacja
Zespół Stevensa-Johnsona (SJS) i zespół toksycznej nekrolizy (TEN) są uważane za ciągłe spektrum ciężkich niepożądanych reakcji skórnych (SCARs – Severe Cutaneous Adverse Reactions). Głównym kryterium różnicującym jest procent odwarstwienia naskórka:
- SJS: zajmuje mniej niż 10% całkowitej powierzchni ciała.
- SJS/TEN overlap: zajmuje od 10% do 30% całkowitej powierzchni ciała.
- TEN: zajmuje ponad 30% całkowitej powierzchni ciała.
Często mylony z nimi jest Rumień wielopostaciowy (EM), który choć również jest schorzeniem skóry charakteryzującym się zmianami tarczowatymi, ma zazwyczaj łagodniejszy przebieg, inną etiologię (częściej infekcyjną) i rzadziej prowadzi do rozległego odwarstwienia naskórka i zajęcia błon śluzowych w tak dramatyczny sposób. Zaburzenia dermatologiczne w SJS są znacznie poważniejsze i wymagają natychmiastowej interwencji medycznej.
Etiologia zespołu Stevensa-Johnsona
W przeważającej większości etiologia zespołu Stevensa-Johnsona jest związana z ekspozycją na leki. Szacuje się, że leki są odpowiedzialne za około 80% przypadków SJS/TEN. Najczęściej do wystąpienia zespołu Stevensa-Johnsona prowadzą:
- Antybiotyki: zwłaszcza sulfonamidy (np. kotrimoksazol), penicyliny, cefalosporyny.
- Leki przeciwdrgawkowe: karbamazepina, fenytoina, lamotrygina, fenobarbital.
- Niesteroidowe leki przeciwzapalne (NLPZ): szczególnie oksykamy (np. piroksykam, meloksykam), ale także ibuprofen, naproksen.
- Allopurynol: lek stosowany w leczeniu dny moczanowej.
- Newirapina: lek antyretrowirusowy.
- Paracetamol: w bardzo wysokich dawkach.
Inne możliwe przyczyny, choć znacznie rzadsze, to infekcje (np. Mycoplasma pneumoniae, wirusy opryszczki, wirusy grypy, HIV), szczepienia, toczeń rumieniowaty układowy, a także idiopatyczne przypadki, gdzie przyczyna pozostaje nieznana. Istnieją również dowody na genetyczne predyspozycje do zachorowania, zwłaszcza w kontekście niektórych leków i specyficznych antygenów zgodności tkankowej (HLA).
Patogeneza SJS/TEN jest złożona i obejmuje aktywację układu odpornościowego, prowadzącą do masowej apoptozy keratynocytów (komórek naskórka). Jest to proces immunologicznie mediowany, w którym komórki cytotoksyczne T odgrywają kluczową rolę w niszczeniu komórek naskórka.
Jak się objawia? Objawy kliniczne
Przebieg zespołu Stevensa-Johnsona jest zazwyczaj gwałtowny i postępujący. Początek choroby często jest poprzedzony objawami prodromalnymi, które trwają od 1 do 14 dni. Do tych objawów należą:
- Gorączka (często wysoka, powyżej 39°C)
- Bóle głowy, mięśni i stawów
- Drażliwość, ogólne złe samopoczucie
- Zaczerwienienie oczu, ból gardła, kaszel
Po fazie prodromalnej pojawiają się charakterystyczne objawy kliniczne na skórze i błonach śluzowych.
1. Zmiany skórne: Początkowo na tułowiu, twarzy i proksymalnych częściach kończyn pojawiają się nieregularne, plamiste wykwity rumieniowe, które szybko rozprzestrzeniają się i zlewają. Są to zmiany zapalne, które w ciągu godzin lub dni przekształcają się w pęcherze i pęcherzyki. Skóra staje się bolesna i nadwrażliwa. Charakterystyczne jest odwarstwianie się naskórka, które przypomina zespół oparzonej skóry. Uciśnięcie i przesunięcie skóry powoduje jej łatwe oddzielanie się, co znane jest jako Objaw Nikolskiego – bardzo ważny w diagnostyce różnicowej. Obszary odwarstwionej skóry są żywoczerwone, wilgotne i bardzo bolesne, co sprawia, że pacjenci z SJS wyglądają jak ofiary rozległych oparzeń.
2. Zajęcie błon śluzowych: Jest to jeden z najbardziej charakterystycznych i bolesnych zespół objawów klinicznych w SJS, występujący u ponad 90% pacjentów.
- Jama ustna: rozległe nadżerki, owrzodzenia i pęcherze na błonie śluzowej jamy ustnej, wargach, języku i dziąsłach. Prowadzi to do silnego bólu, trudności w połykaniu (dysfagia) i mówieniu, ślinotoku. Usta często pokryte są krwawymi strupami, co uniemożliwia przyjmowanie pokarmów i płynów.
- Oczy: zapalenie spojówek, owrzodzenia rogówki, światłowstręt, ból, ropna wydzielina. Nieleczone mogą prowadzić do poważnych powikłań, w tym ślepoty.
- Narządy płciowe: bolesne nadżerki i owrzodzenia na błonie śluzowej cewki moczowej, pochwy, jąder. Prowadzi to do bolesnego oddawania moczu (dyzuria).
- Drogi oddechowe: zajęcie błony śluzowej tchawicy i oskrzeli może prowadzić do kaszlu, duszności, niewydolności oddechowej.
- Przewód pokarmowy: rzadziej, ale mogą pojawić się wrzody w przełyku, żołądku, jelitach, powodujące krwawienia.
3. Objawy ogólnoustrojowe: Pacjenci z SJS są w ciężkim stanie ogólnym. Często występują:
- Sepsa (posocznica) – główna przyczyna zgonów.
- Zaburzenia elektrolitowe i odwodnienie, w wyniku utraty płynów przez uszkodzoną skórę.
- Niewydolność nerek, wątroby, płuc.
- Pankreatopatia.
- Zmiany neurologiczne (drgawki, encefalopatia).
Przebieg choroby jest dynamiczny. Zmiany mogą rozwijać się bardzo szybko, a stan pacjenta pogarszać się z godziny na godzinę, co wymaga natychmiastowej hospitalizacji i intensywnej opieki. Każdy przypadek zespołu Stevensa-Johnsona jest nagłym stanem medycznym.
Rozpoznanie zespołu Stevensa-Johnsona
Rozpoznanie zespołu Stevensa-Johnsona opiera się przede wszystkim na obrazie klinicznym i wywiadzie dotyczącym ekspozycji na leki. Kluczowe jest szybkie rozpoznanie i wycofanie podejrzanego leku.
Narzędzia diagnostyczne:
- Wywiad medyczny: Dokładne zebranie informacji o wszystkich przyjmowanych lekach (zarówno tych na receptę, jak i bez recepty, suplementach) w ciągu ostatnich 1-4 tygodni przed wystąpieniem objawów.
- Badanie fizykalne: Ocena charakterystycznych zmian skórnych, obecności pęcherzy, nadżerek, odwarstwień naskórka i szczególnie zajęcia błon śluzowych. Sprawdzenie Objawu Nikolskiego.
- Biopsja skóry: Jest kluczowa dla potwierdzenia rozpoznania. Obraz histopatologiczny pokazuje martwicę keratynocytów w pełnej grubości naskórka, oddzielenie naskórka od skóry właściwej oraz skąpy naciek zapalny. Pozwala to na różnicowanie z innymi chorobami pęcherzowymi i chorobami skóry, takimi jak autoimmunologiczne choroby pęcherzowe czy gronkowcowy zespół oparzonej skóry, który ma odmienną etiologię i leczenie.
- Badania laboratoryjne: Charakterystyczne mogą być leukocytoza, niedokrwistość, zwiększone markery stanu zapalnego (CRP, OB), zaburzenia elektrolitowe, podwyższone poziomy mocznika i kreatyniny (wskazujące na uszkodzenie nerek) oraz enzymów wątrobowych (wskazujące na uszkodzenie wątroby). Posiewy krwi i skóry są konieczne w przypadku podejrzenia sepsy.
W ocenie ciężkości i rokowania stosuje się Skalę SCORTEN. Jest to system punktowy, który ocenia ryzyko zgonu na podstawie siedmiu parametrów: wieku, częstości akcji serca, współistniejących nowotworów, zajętej powierzchni ciała, stężenia mocznika, glukozy i wodorowęglanów w surowicy. Wyższy wynik w Skali SCORTEN oznacza gorsze rokowanie.
Jak wygląda leczenie?
Leczenie zespołu Stevensa-Johnsona jest złożone, wymaga natychmiastowej interwencji i zazwyczaj odbywa się w warunkach intensywnej terapii (OIOM) lub specjalistycznych oddziałach oparzeniowych. Celem leczenia jest przede wszystkim usunięcie czynnika wywołującego, intensywne leczenie wspomagające i profilaktyka powikłań.
1. Natychmiastowe odstawienie leku: To absolutny priorytet. Należy bezzwłocznie odstawić wszystkie podejrzane leki, niezależnie od stopnia pewności.
2. Intensywna opieka wspomagająca: Jest to podstawa leczenia i w dużej mierze decyduje o przeżyciu pacjenta.
- Opieka nad skórą: Rany skórne traktuje się jak oparzenia II stopnia. Konieczne są codzienne, aseptyczne zmiany opatrunków. Stosuje się jałowe opatrunki, maści z antybiotykami lub inne specjalistyczne preparaty (np. maść z sulfadiazyną srebra), które wspomagają gojenie i zapobiegają infekcjom. Regularne kąpiele z łagodnymi środkami antyseptycznymi.
- Płynoterapia: Utrata płynów przez uszkodzony naskórek jest znaczna, dlatego kluczowe jest intensywne nawadnianie dożylne w celu utrzymania równowagi wodno-elektrolitowej i zapobiegania wstrząsowi.
- Żywienie: Z powodu bolesnych zmian w jamie ustnej, pacjenci często nie są w stanie przyjmować pokarmów doustnie. Konieczne jest żywienie dojelitowe przez sondę nosowo-żołądkową lub całkowite żywienie pozajelitowe.
- Leczenie bólu: Zmiany zapalne i odwarstwiona skóra są niezwykle bolesne. Konieczne jest agresywne leczenie bólu za pomocą silnych analgetyków (opioidów).
Opieka nad błonami śluzowymi:
- Oczy: Należy regularnie przemywać oczy solą fizjologiczną, stosować maści nawilżające, a w ciężkich przypadkach krople z kortykosteroidami lub cyklosporyną. Niezbędna jest konsultacja okulistyczna.
- Jama ustna: Regularne płukanie jamy ustnej roztworami antyseptycznymi (np. chlorheksydyną) i środkami miejscowo znieczulającymi.
- Narządy płciowe: Dokładna higiena i stosowanie maści łagodzących.
- Zapobieganie infekcjom: Uszkodzona bariera skórna i upośledzona odporność predysponują do zakażeń bakteryjnych, zwłaszcza sepsy. Stosuje się ścisłą higienę, ale profilaktyczne podawanie antybiotyków nie jest zalecane. Antybiotyki podaje się tylko w przypadku potwierdzonej infekcji lub sepsy.
- Monitorowanie funkcji narządów: Ciągłe monitorowanie parametrów życiowych, funkcji nerek, wątroby, płuc i elektrolitów.
3. Leczenie farmakologiczne (specyficzne): Skuteczność specyficznego leczenia farmakologicznego w SJS/TEN jest przedmiotem debat i badań.
- Kortykosteroidy ogólnoustrojowe: Ich zastosowanie jest kontrowersyjne. Niektóre badania sugerują, że wczesne podanie dużych dawek kortykosteroidów może zahamować przebieg choroby, ale inne wskazują na zwiększone ryzyko infekcji i opóźnienie gojenia. Obecnie większość ekspertów odradza ich rutynowe stosowanie.
- Immunoglobuliny dożylne (IVIg): IVIg teoretycznie mogą neutralizować czynniki odpowiedzialne za apoptozę keratynocytów. Ich skuteczność jest bardziej udokumentowana w przypadku zespołu toksycznej nekrolizy (TEN) niż SJS, ale również jest tematem dyskusji.
- Cyklosporyna: To lek immunosupresyjny, który jest coraz częściej rozważany jako opcja terapeutyczna, zwłaszcza we wczesnej fazie choroby. Badania sugerują, że może zmniejszać śmiertelność i przyspieszać reepitelizację.
- Etanercept: Lek biologiczny, inhibitor TNF-alfa, badany w niektórych ośrodkach.
- Plazmafereza: Procedura, która ma na celu usunięcie z krwi krążących mediatorów zapalnych. Stosowana w ciężkich, opornych na inne terapie przypadkach.
Kluczowe jest, aby leczenie było prowadzone przez zespół multidyscyplinarny, w skład którego wchodzą dermatolodzy, intensywiści, okuliści, chirurdzy plastyczni (oparzeń) oraz dietetycy.
Powikłania i rokowanie
Zespół Stevensa-Johnsona wiąże się z poważnymi powikłaniami i wysoką śmiertelnością, która wynosi około 5-15% dla SJS, a dla zespołu toksycznej nekrolizy dochodzi do 30-50%. Główne przyczyny zgonów to sepsa, niewydolność wielonarządowa i powikłania oddechowe.
Nawet po wyzdrowieniu pacjenci mogą cierpieć na długoterminowe konsekwencje, takie jak:
- Oczne: przewlekłe zapalenie spojówek, suchość oka, światłowstręt, blizny na rogówce, a nawet ślepota.
- Skórne: przewlekłe bliznowacenie, zaburzenia pigmentacji (hipo- lub hiperpigmentacja), nadmierne rogowacenie, suchość skóry.
- Oddechowe: zwężenie dróg oddechowych, przewlekła obturacyjna choroba płuc.
- Inne: zwężenie przełyku, pochwy, cewki moczowej, przewlekłe grzybice paznokci, przewlekłe zmęczenie.
Każdy przypadek zespołu Stevensa-Johnsona wymaga długotrwałej obserwacji i rehabilitacji.
Podsumowanie
Zespół Stevensa-Johnsona (SJS) to rzadka, ale niezwykle groźna choroba skóry i błon śluzowych, która wymaga natychmiastowego rozpoznania zespołu Stevensa-Johnsona i intensywnego leczenia. Jej etiologia zespołu Stevensa-Johnsona jest w większości przypadków związana z lekami, co podkreśla znaczenie ostrożności w farmakoterapii i świadomości pacjentów o potencjalnych reakcjach. Objawy kliniczne są dramatyczne, przypominające rozległe oparzenia, a przebieg zespołu Stevensa-Johnsona jest dynamiczny i może szybko prowadzić do niewydolności wielonarządowej i sepsy. Kluczowe dla poprawy rokowania jest szybkie odstawienie leku wywołującego oraz agresywne leczenie wspomagające w specjalistycznym ośrodku, często w warunkach intensywnej terapii. Mimo postępów w medycynie, zespół toksycznej nekrolizy i SJS pozostają wyzwaniem terapeutycznym, a ich powikłania mogą mieć długotrwały wpływ na jakość życia pacjentów.
Źródła:
Materiał ten ma charakter wyłącznie informacyjny i edukacyjny. Nie należy go traktować jako jedynej podstawy do podejmowania decyzji. W przypadku wątpliwości lub przed podjęciem konkretnych działań, zawsze zaleca się konsultację z lekarzem lub farmaceutą.